




«Kvazar» is an open multiprocessor software complex of molecular modeling, which combines the advantages of the most successful modern molecular modeling package (support for high-performance computing, cross-platform graphical user interface, a large variety of theoretical methods and approaches), but at the same time having a flexible, easily modifiable architecture.
https://www.sgu.ru/structure/nano-bio/modelling/mathematicalsimulation/proekt-kvazar
Possibilities of software package
Program complex «Kvazar» contains a wide set of tools allows researchers to solve the specific scientific problem at all its stages: a design of geometry of the objects; the task of periodic boundary conditions and external conditions; the numerical experiments using a variety of mathematical models; preservation and visualization of the results. These tools focus on complex multiprocessor systems, and work on a personal computer.
Developing complex of molecular modeling is used to solve a wide class of problems in biophysics, medicine and nanoelectronics:
- The calculation of emission, electron energy and strength characterizes of carbon nanostructures, including the defect-free single-walled and multi-walled nanotubes, graphene and its modifications, the nanotube of complex shape, fullerenes, composite materials, in order to create models of the improved versions of electronic nanodevices on their basis;
- The calculation of the electrical conductivity and the thermal conductivity of nanostructures and biosystems to create the high sensitivity sensor devices on their basis;
- The modeling of physical and chemical processes in the intima of arteries at the atomic and molecular-cellular level to identify the mechanism of the lipoproteins penetration in interendothelial space;
- The modeling of the transport of organic and inorganic molecules on graphene in order to identify the most effective approach to manage their movement;
- The modeling of the interaction between the components of molecular complexes of biomolecules and carbon nanostructures to develop new devices of bioelectronics based on them;
- The modeling of the self-assembly processes for biological macromolecules in order to develop the modern technologies to produce of bionanomaterials.
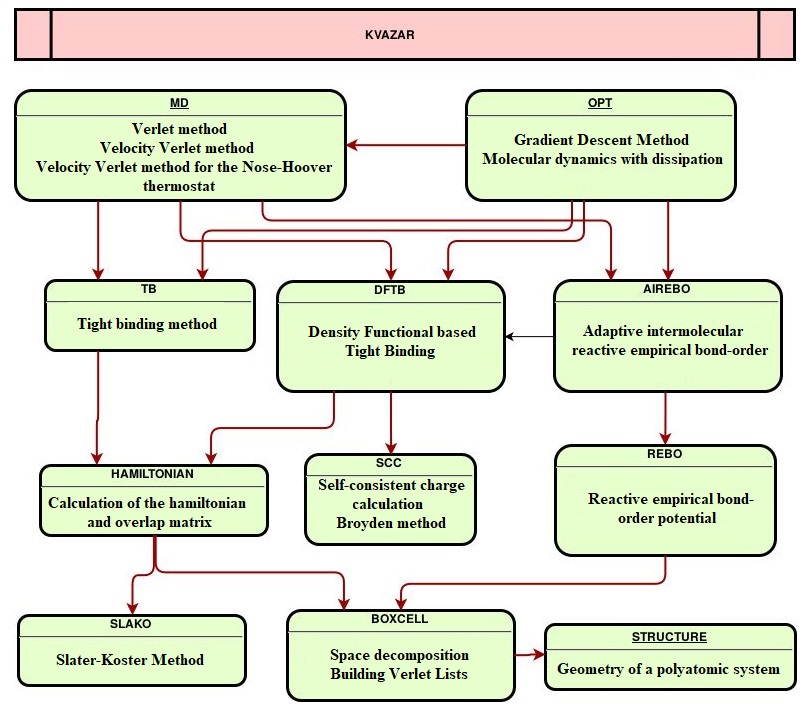
This complex implemented the classical approaches of molecular modeling, the original mathematical models and new versions of existing methods of mathematical modeling to solve the above problems:
- classical molecular dynamics with algorithms of Berendsen and Noze-Hoover thermostats
- modified molecular mechanical method based on the Brenner potential;
- molecular mechanical method based on the REBO potential;
- molecular mechanical method based on the capacity of AIRBEO potential;
- the adapted quantum-chemical tight binding method for the study of electronic and mechanical properties of carbon nanostructures;
- the mathematical hybrid model of QM/MM (Quantum Mechanics/Molecular Mechanics) with an original algorithm to determine the boundaries of the active region based on the calculation of local stresses of atomic grid;
- the original method for calculating the local stresses of atomic grid;
- the coarse model using MARTINI force field;
- the classical molecular mechanical method using the potential AMBER.
The developed software system is focused on the use of the hybrid parallel architecture that combines different technologies of parallel programming (MPI) and various types of calculators (CPU and GPU), which allows several times to reduce the simulation of physical and chemical processes at the atomic and molecular level.
Results of approbation of methods and approaches to be implemented in the software complex Kvazar, are presented in the following main publications of the research group : http://nanokvazar.ru/osnovnye-publikatsii

